[프레스나인] 유럽연합(EU)이 도입한 임상시험규정(Clinical Trial Regulation, CTR)에 대한 유예기간 종료가 다가오면서 유럽에서 임상을 준비하는 국내 제약·바이오 기업들도 달라진 제도에 대한 모니터링이 필요해 보인다.
EU는 임상시험 정보에 대한 투명성과 접근성 향상을 목표로 올해부터 CTR 제도를 도입했다. 유예기간은 오는 2023년 1월31일까지다.
기존까지 유럽 임상은 유럽의약품청(EMA) 가이드라인에 따라 유럽 각 국가별로 승인을 받아 진행했다. 향후에는 임상시험 정보 시스템(CTIS)에 작성한 레지스트리를 토대로 동시 승인신청을 할 수 있다.
2025년부터는 기존 제도(Clinical Trials Directive, CTD)로 진행 중인 임상도 이 CTIS에 임상 데이터를 등록해야 한다.
제도 도입 핵심 취지가 임상 데이터에 대한 투명한 공개인 만큼 기업들은 기존보다 더 자세한 임상 프로토콜, 디자인을 공개해야 한다.
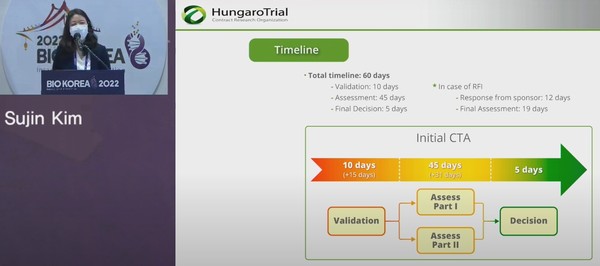
유럽 임상시험 수탁기관(CRO)인 헝가로트라이얼(HungaroTrial) 김수진 이사(비즈니스개발)는 지난 11일 한국보건산업진흥원 주최로 서울 코엑스에서 열린 '바이오코리아 2022' 발표를 통해 최종 승인까지 명확한 타임라인이 EU CTR의 가장 큰 장점이라고 봤다.
김 이사는 "타임라인이 넘어가면 자동으로 승인이 되는 시스템"이라며 "현실적으로 많지 않지만 아무런 보완의견이 없을 때 기준으로 60일, 추가 자료제출 요구가 있다면 한 달 정도 추가가 되서 최장 90일정도가 타임라인"이라고 설명했다.
임상시험계획(IND) 승인 과정에선 필수문서 제출여부, 제출문서 완성도, 임상 도즈설정, 독성시험, 안전성 근거 문헌 관련, 이상반응 보고, 시험대상자에 대한 피해 최소화 데이터 분석 등평가항목에서 보완과 반려가 많다고 설명했다.
또한 미국 식품의약국(FDA) 임상시험 용어를 사용하는 한국과 달리 유럽에선 IND를 'CTA(Clinical Trial Application)', 임상시험심사위원회(IRB)를 'EC(Ethic Committee)', 규제당국(RA)을 'CA(Competent authority)' 신약승인신청(NDA)를 'MAA(Marketing Authorisation Application)' 등으로 지칭한다고 김 이사는 전했다.